
Compliance Overpowers Competition
The US Food and Drug Association (USFDA) keeps a close oversight of the pharmaceutical industry. Stringent audits and rigorous approvals signify a high measure of confidence in patients about the pharma products. However, for companies responsible for manufacturing operations, FDA audits can inspire worry. The regulations and compliance requirements must find a balance against a company’s sustainability goals in the competitive global market which is highly price sensitive.
The Surprise Element / FDA Surprises are Not Pleasant
With significant preparation required for FDA audits, they are often intimated well before time, but not always. While routine surveillance inspections are notified, the for-cause inspections are mostly unannounced.
Regulatory compliance is an unsaid component for the operations of life sciences and pharma businesses. Companies are bound to abide by certain guidelines as mentioned in the current good manufacturing practices (cGMP) standards. However, with the increase in global competition, there is a tendency to outperform in terms of financial growth. This often leads to manipulation, ignorance, and non-disclosures.
To fulfill its designated purpose of having a safe and regulated environment for patients, the FDA needs to step in. With increasing defaults and manipulations, the regulatory body has increased its surprise visits to manufacturing facilities. This calls for enterprises to be prepared by maintaining best practices and higher quality standards.
The Dreaded 483s / 483s – The Scary Number
Audits are mandatory and dreadful irrespective of the company’s preparedness. During an audit, if the FDA finds that a pharmaceutical manufacturing unit is non-compliant with any part of the required standards as per cGMP, the agency takes action. The actions can range from coaching the firm to closing the facility including many corrective actions between these extremities.
483 – is the most dreaded number that any regulated company would want to see. FDA issues a 483, also called a warning letter, that lists all the violations inspectors find during their audits and the steps to be taken to address them. In 2022, the FDA issued at least 466 Form 483s were issued, 231 (or 49%) of which cited 21 CFR 211.
After receiving a 483, companies need to deck up for responses. There is a list of findings, follow-up guidelines, a line of action along with a deadline. Dealing with 483s engages a lot of effort, precious time, and intellect of resources. No doubt, they are a dreaded number!
Top 10 Citations – FDA Data Dashboard:
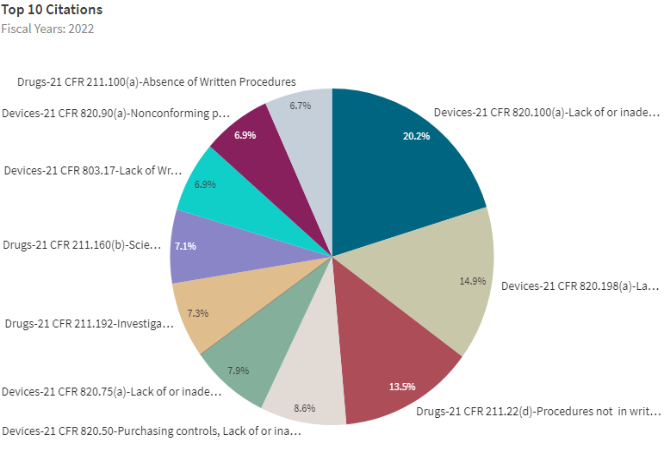
Causes that Triggered Most 483s in 2022:
Top 10 Citations Explained
1. Written Procedure Deviations
Finished products are expected to conform to written procedures mentioned in the production and process control documents. This is necessary to ensure that the finished goods have the quality, strength, and purity they are intended to represent. This conformance must be followed at every stage and functional performance. Any deviations from the written procedures must be justified by the companies.
2. Nonconforming Products
For products that do not conform to specified requirements, manufacturers must maintain and establish control procedures. The evaluation of nonconformance determines the need for an investigation and must be documented. Manufacturers are required to have review and authority procedures for disposing nonconforming products. There is also a need for establishing procedures for rework. To ensure that the product meets its current approved specifications, the nonconforming product must be reevaluated. All the control procedures, evaluation guidelines, rework and retesting specifications must be documented. Any missing adherence to the control procedures follows objections from the regulatory bodies.
3. Medical Device Reporting Procedures
All the manufacturers of medical devices are obliged to develop, maintain, and implement written procedures pertaining to the internal systems, documentation, and recordkeeping. This includes effective identification, communication, evaluation, and review of events. Manufacturers must document information about a reportable event along with all medical device reports. They are also required to maintain documentation for systems that ensure access to information for timely follow ups and inspection by FDA.
4. Laboratory Controls
Every laboratory process including sampling plans, test procedures, control mechanisms, including any change in these at any point of time must be reviewed and approved by the quality control unit. Any deviations from these written specifications and other laboratory controls must be recorded and justified. Laboratory controls are defined to assure that drug containers, components, closures, labeling, and in-process materials conform to appropriate quality, purity, identity, and strength standards. The standard also specifies that the calibration schedules of instruments, gauges, and other devices must be specifically documented, and their accuracy limits be defined. In case of any deviations with regards to the above, the FDA can raise a concern.
5. Production Record Review
According to this guideline, all drug product production and control records must comply with all the approved written procedures before a batch is released. Any unexplained discrepancies or failure of a batch to meet its specifications can rightfully be investigated. This investigation can further be extended to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy.
6. Quality System Process Validation
This guideline mandates quality system regulation. When the result of a particular process cannot be verified, the process needs to be validated and approved with a high degree of assurance. Under this section, manufacturers are expected to establish procedures for monitoring and control of process parameters for validated processes. Process validation can only be done by qualified individual(s). It also emphasizes the monitoring and control methods, data, and other important considerations such as the date of validation, major equipment used, and individual(s) involved. For any deviation that occurs, reevaluation and revalidation of the processes must be conducted.
7. Purchasing Controls (Supplier Management)
To adhere to quality specifications, manufacturers are required to maintain specific procedures to ensure that all the purchased products and services conform to those specifications. This guideline states that manufacturers must evaluate and select consultants, suppliers, and contractors based on their ability to meet specified requirements. Also, it mandates maintaining records of acceptable consultants/suppliers/contractors and defining the extent of control to be exercised over them based on the evaluation results. All the purchasing data needs to be documented in detail. The data must also speak of notifying the manufacturer of any change in the product or service so that manufacturers may determine whether the changes may affect the quality of a finished device.
8. Corrective and Preventive Action
FDA has predefined guidelines for implementing corrective and preventive actions. This starts with analyzing processes, reports, complaints, returned product, and other quality data to identify potential causes of the nonconforming product. A thorough investigation is followed by identifying, verifying, and validating the corrective and preventive action to ensure its effectiveness. In addition, the changes in methods and procedures must ensure they do not adversely affect the finished product. All the activities and relevant information on identified quality problems must be submitted for management review and well-documented.
9. Records – Complaint Files
Maintenance of complaint files is an important consideration. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit. There are specific guidelines for processing complaints. Manufacturers are required to have procedures for receiving, reviewing, and evaluating complaints. They must evaluate complaints to determine whether an investigation is necessary. If the complaint is related to possible failure of a device, labeling, or packaging to meet any of its specifications, it shall be reviewed, evaluated, and investigated. Complaints representing an event that must be reported to FDA under part 803 of 21 CFR must be promptly acted upon.
10. Responsibilities of Quality Control Unit
FDA demands every manufacturer must have a quality control unit that shall have the responsibility and authority to approve or reject all components including drug product containers, packaging materials, in-process materials, labeling, and the authority to review. The unit shall be responsible for rejecting or approving drug products processed, manufactured, packed, or held under contract by another company. Additionally, it can also exercise its rights to approve or reject procedures or specifications impacting on the identity, strength, quality, and purity of the drug product. These responsibilities and procedures applicable to the quality control unit must be in writing and such written procedures shall be followed.
“Caliber has helped us maintain high quality standards and address FDA concerns. Their conscious effort and highly resilient software established a healthy ecosystem for us.”
Waking Up to A 483-free Morning Everyday
FDA 483s are on a manufacturer’s subconscious mind, ALWAYS. The undeniable component of quality assurance and adherence to the regulations make it more prevalent. While the major reasons for 483 citations are elaboratively discussed, we understand that most of the violations relate to written procedures and documentation. With stringent timelines to follow while ensuring product efficacy, safety, and quality it becomes difficult to document every single detail manually. A great way to nullify the chances of receiving this undesired correspondence from the FDA is to digitize quality processes – documentation, quality assurance, and learning management.
The FDA promotes modernization of health care product manufacturing. There are several steps taken by the regulatory agency to stimulate advanced manufacturing. They have also emphasized how integrated quality management drives pharmaceutical success. Today, digital technologies are enabling regulated companies to achieve phenomenal quality resilience by eliminating paper, avoiding production bottlenecks, and thereby preventing warning letters.
Avoid Corrections, Take Preventions
Proactive actions outweigh reactive mechanisms in many ways. It saves energy, time, and costs too. No company in the life sciences industry willfully rebels against cGMP. Companies receive 483s because something was overlooked. Mostly non-compliance issues are a result of lack of collaboration, broken workflows, or simply human errors. By having a clarity of purpose and digitalized quality approach many of these FDA warning letters can be prevented. Quality-driven operations promise increased efficiency and successfully negates FDA allegations.
Integrated Approach to Quality is the Success Mantra
When quality is spread across departments, it makes more sense. It is more efficient, cost-effective, and scalable. Instead of mapping quality in batches to specific areas, having an integrated system where everything is inter-linked to quality and each other too, will give a better result. Overall digitalization brings in significant enhancement to processes, products, and people.
Caliber’s epiq – Enterprise Platform for Integrated Quality has recently garnered the attention of regulated companies in the life sciences industry. The product has brought in a revolution with a 3-in-1 solution for quality management. It has quality assurance, documentation management, and learning management under one roof which makes it an overall solution to a company’s digital quality needs.
FAQs


