
The U.S. Food and Drug Administration (FDA) issued a draft guidance in March 2026 outlining its current thinking on the content and format of Form 483 responses. This document is currently being distributed for comments only and does not establish enforceable responsibilities.
It offers useful insight into how the FDA may evaluate responses and what typically characterizes a strong, well-supported reply.
Why this draft guidance matters
Until now, many manufacturers have followed internal practices, consultant advice, and industry norms to structure their responses. The draft guidance does not replace statutory or regulatory requirements, but it does provide helpful direction on:
- How companies may organize their written response
- What types of information can strengthen the response
- How FDA reviewers may look at investigations, CAPA, and risk assessments
A Form 483 is not just a list of observations. It is also an opportunity to demonstrate that the company:
- Understands the observation and its context
- Has thoroughly investigated the issue
- Is implementing appropriate, evidence-based CAPA
- Operates a quality system capable of preventing recurrence
Weak responses are a major reason the FDA escalates to warning letters. The draft guidance gives more visibility into how the agency may view the adequacy of a response.
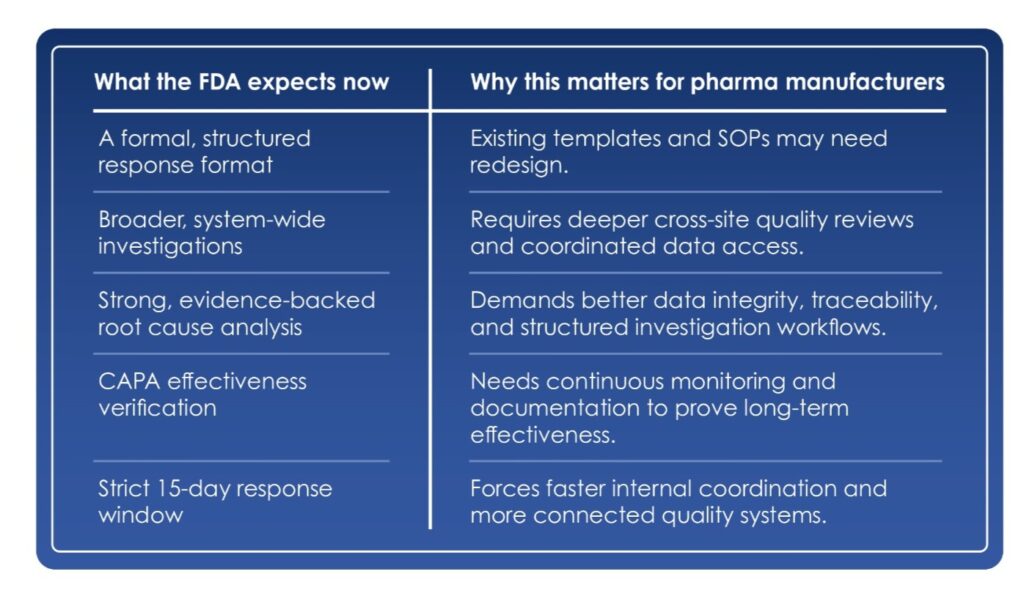
The 15‑day window is non-negotiable
The draft guidance reinforces FDA’s long‑standing practice around timelines. In general, manufacturers have 15 business days to submit a written response.
Responses submitted within this window allow time for the FDA to decide on the effectiveness of action taken to address the observations. Quality leaders would need to:
- Start investigations immediately
- Bring quality, manufacturing, and cross‑functional teams together quickly
- Move from observation to root cause in days, not weeks
- Ensure data, documents, and evidence are easy to access
This can be challenging for companies relying on manual or disconnected systems, indirectly highlighting the value of having accessible, reliable data.
What a strong response looks like
The draft guidance suggests a structured approach to responses. Companies may find it useful to organize their responses to include:
- A clear executive summary
- Investigation findings
- Evidence-based root cause analysis
- Detailed CAPA
- Product and patient risk assessments
- Supporting documentation and timelines
The document also notes that unstructured, narrative-only responses can be harder to evaluate. A clear, logical structure may make it easier for reviewers to see how the company has addressed each observation.
Investigate beyond the single observation
The draft guidance highlights the importance of evaluating the broader system, and not just individual observation.
The FDA encourages companies to consider whether the issue affects:
- Other batches or products
- Other processes
- Other manufacturing facilities
- Contract partners
If a response addresses only the single observation, the reviewers may question whether the quality system can reliably identify and manage broader risks.
Root cause and CAPA: show the evidence
The draft guidance also cautions against narrowing in too quickly on the first suspected cause or testing only to confirm a favored hypothesis. Instead, companies are encouraged to:
- Considered multiple possible causes
- Used data to support their conclusion
- Verified CAPA effectiveness with measurable evidence
Closing CAPA without supporting data can increase the likelihood of repeat observations, re-inspection, or additional regulatory attention.
What this means for pharma quality leaders
This may prompt manufacturers to:
- Review and refine internal
- Form 483 response templates and SOPs
- Strengthen investigation workflows and cross-site collaboration
- Improve access to reliable, inspection-ready data
- Formalize how product and patient risks are assessed and documented
- Build greater discipline around CAPA effectiveness checks
For many organizations, this also sparks a broader question: Can our current systems support the speed, structure, and evidence that a strong response requires?
Many organizations use connected quality platforms to help teams accelerate investigations, maintain clear traceability, and stay inspection-ready always.
If you’re rethinking how your organization handles investigations, CAPA, and inspection responses, explore how Caliber’s connected quality solutions can support faster, more robust, and compliant responses to FDA inspection observations
Important note
This article summarizes key themes from the FDA’s draft guidance on Form 483 responses. The document is being distributed for comment only and does not establish legally enforceable responsibilities. Companies should continue to rely on applicable regulations, internal procedures, and official FDA communications when preparing responses. An effective action to address most data integrity, compliance, and quality issues is to digitalize your internal systems like LIMS, QMS, EBR, etc. Speak to a Caliber Expert now to seek support if you are facing a 483.
Explore how Caliber’s connected quality solutions support faster, compliant responses to FDA inspection observations.
FAQs


